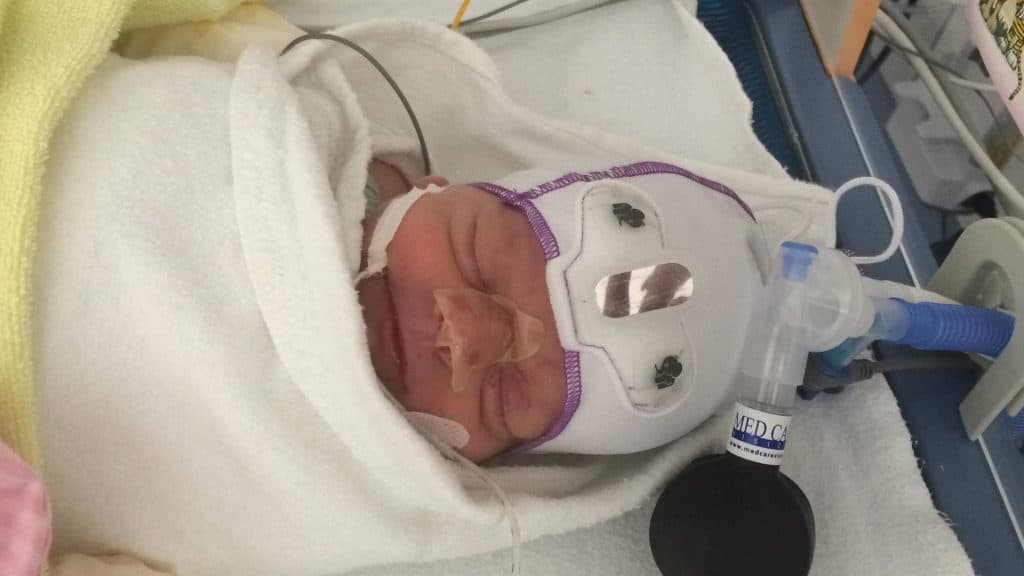
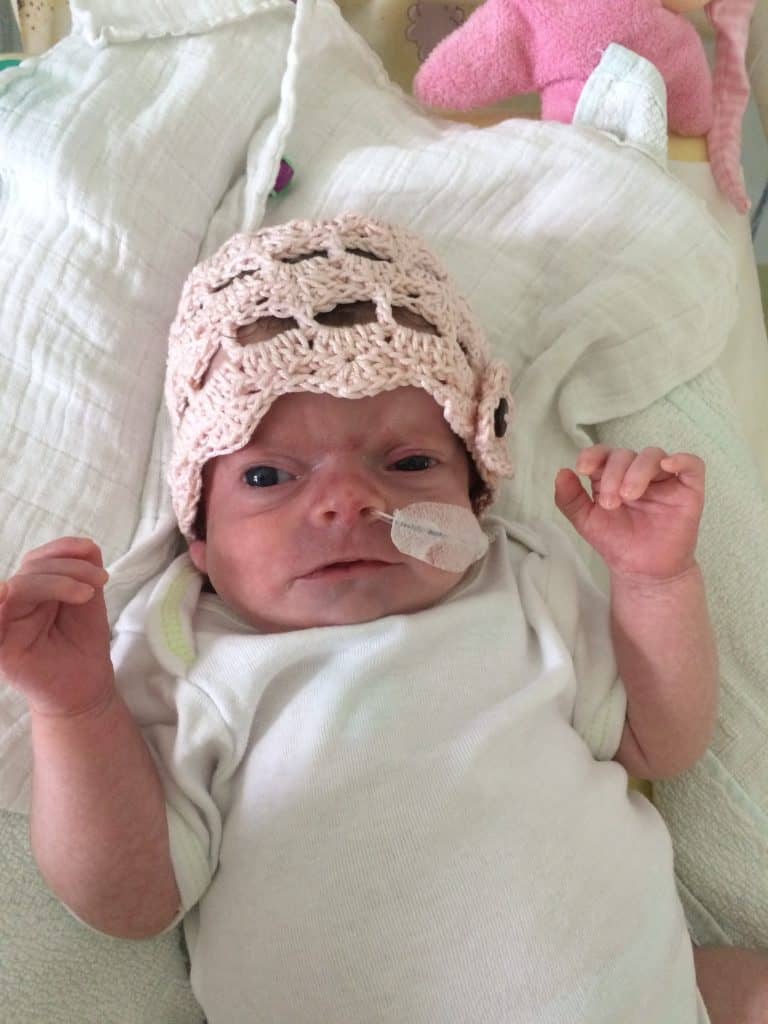
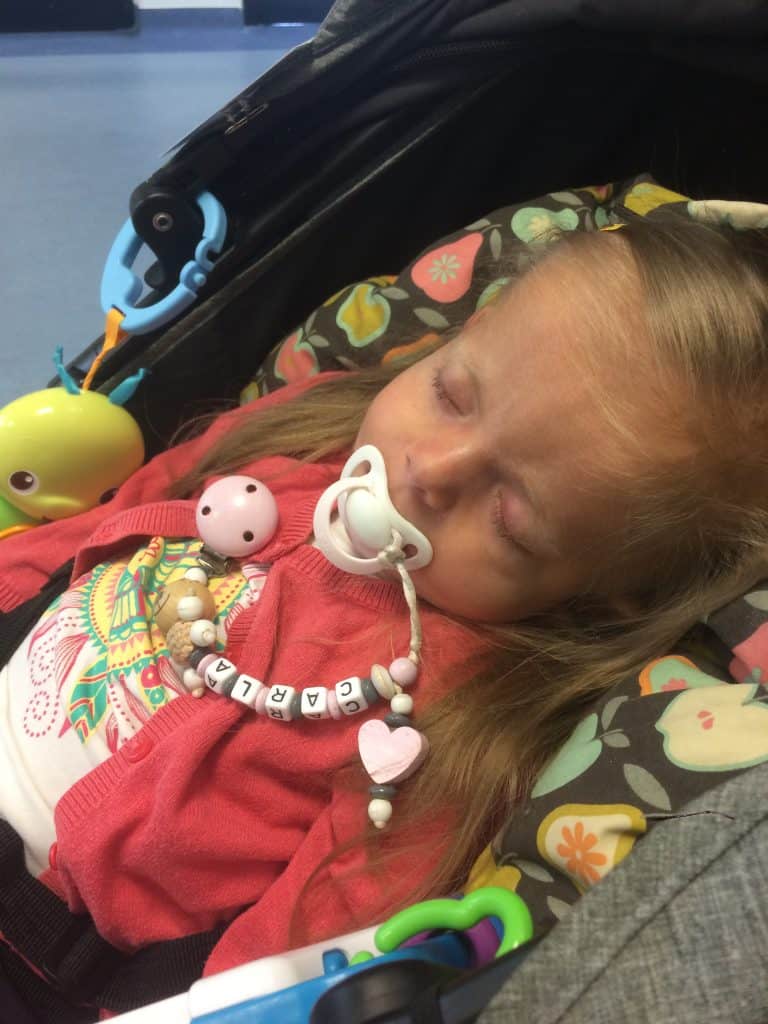
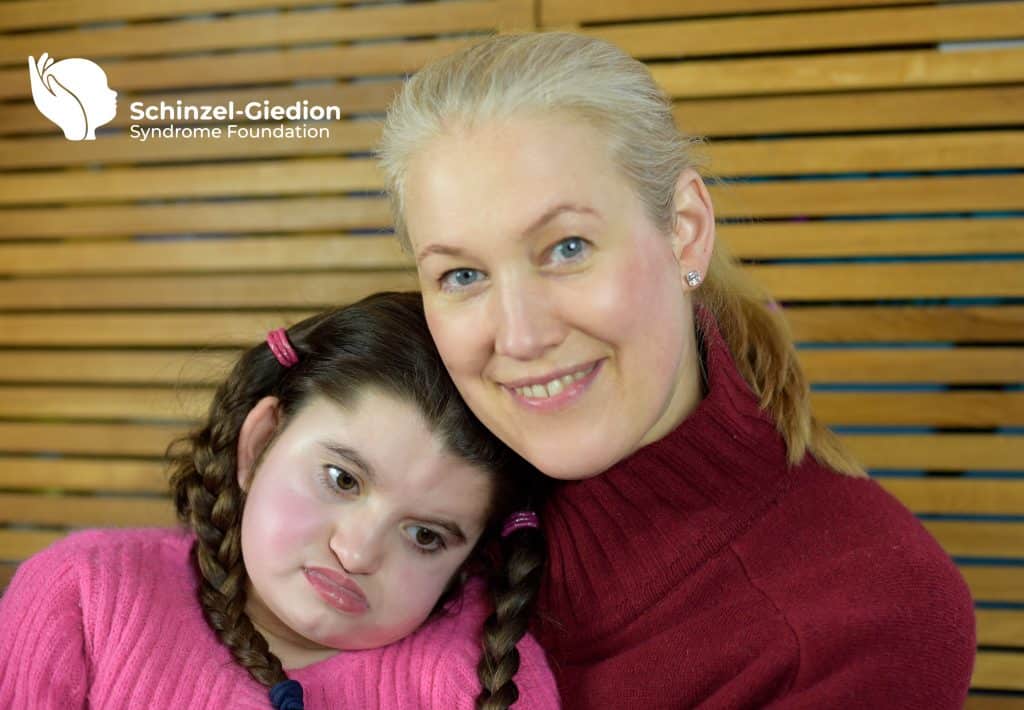The Schinzel-Giedion Syndrome Foundation is a member of COMBINEDBrain
In January 2020 The Schinzel-Giedion Syndrome Foundation joined COMBINEDBrain, a non-profit consortium of 25 patient-advocacy groups, each representing a different rare genetic neurodevelopmental disorder. COMBINEDBrain’s mission is to speed clinical trial readiness for severe cognitive disorders by pooling resources and working together across all of our member disorders.
The Schinzel-Giedion Syndrome (SGS) Foundation is represented by Nuala Summerfield, Founder and Chair. The organisation’s mission is to provide support to families caring for a child with SGS, to raise awareness of SGS and to facilitate and support medical research that will help find better treatments to improve the quality and length of life of children living with SGS. The Schinzel-Giedion Syndrome Foundation is a volunteer, parent-led organisation and represents the international SGS community.
We are excited and honoured to be a member of COMBINEDBrain and to have the opportunity to work alongside so many other like-minded organisations, all focused on treating and curing ultra-rare neurodevelopmental disorders. Through this collaboration, we are able to share ideas, resources and contacts. We learn from and inspire each other and work together to find strategic partnerships. Most importantly, our membership in COMBINEDBrain helps spread much-needed awareness about Schinzel-Giedion Syndrome (SGS) and SETBP1 mutations and gives us access to biopharma companies that can help us achieve our goal of finding targeted treatments for children with SGS.
COMBINEDBrain was founded by Dr. Terry Jo Bichell, whose adult son has Angelman Syndrome. In 2009, Terry Jo decided to go back to school, earning her PhD in neuroscience in order to work towards finding a cure for her son. She created COMBINEDBrain in 2019 to share her experience with the next wave of rare diseases. She’s a force to be reckoned with in the rare disease community, and we are so grateful to have her as a mentor and friend.
The other members of CB that we are in collaboration with (in reverse alphabetical order) are:
Yellow Brick Road Project (HNRNPH2) is represented by Trish Flanagan, President. The YBRP connects families and drives research forward into HNRNPH2 mutations to improve these rare patients’ lives. This is a small but fiesty organization who are laser focused on getting to clinical trials, treatments and a cure for the rare X-linked HNRNPH2 related neurro-developmental disorder.
SynGAP Research Fund (SRF) is represented by Mike Graglia, Managing Director & co-founder. SRF’s mission is to support the research and development of treatments, therapies and support systems for SynGAP1 patients worldwide. SRF is entirely parent led and has committed over $1.2M to research since it was created, 100% of donations go directly to support research.
STXBP1 Foundation is represented by Charlene Son Rigby, President. The STXBP1 Foundation’s mission is to raise awareness of STXBP1 disorders, and to accelerate the development of therapies and hopefully a cure for our patients.
SLC6A1 Connect is represented by Amber Freed, CEO & Co-Founder. The mission of SLC6A1 Connect is to cure every person with SLC6A1.
SETBP1 Society is represented by Haley Oyler, President. Their mission is to provide support to individuals with SETBP1 disorder and their families, to promote discussion and fund research, and to bring awareness and education to the public. SETBP1 Society is an internationally-focused volunteer 501(c)(3) organization based in the US with a focus to identify targeted treatments to help individuals impacted by SETBP1 disorder.
SATB2 Gene Foundation is represented by Allison Kaczenski, President & Founder. The SATB2 Gene Foundation was established to enrich the lives of individuals with SATB2-associated syndrome, including those diagnosed with the condition and their families, through support, research and education.
Project 8p Foundation is led by Bina Maniar Shah, President & Founder. Project 8p Foundation is a 501(c)(3) non profit organization established to accelerate the discovery of treatments for chromosome 8p disorders with a translational research program and a standard of care to empower meaningful lives in a unified community today. Chromosome 8p is not just a rare genetic disease, but the many genes and pathways can be clues to common brain-related diseases.
Project Alive is represented by Kim Stephens, DBA, President Their mission is to find and fund a cure for Hunter Syndrome (also known as Mucopolysaccharidosis or MPS II) through research and advocacy. Project Alive is a powerful voice for children and adults with Hunter Syndrome, bringing together families and advocates with researchers, industry, and regulators.
PBD Project is represented by Andrew Longenecker, Founder. Their mission is to PBD Project is to fund medical research with the objective to provide meaningful positive clinical impact for patients with Peroxisome Biogenesis Disorders (PBD), with a focus on Zellweger Spectrum Disorder (ZSD) caused by mutations to PEX10 gene.
NR2F1 Foundation is represented by Carlie Monnier, Board President. The mission of the NR2F1 Foundation is to empower families and individuals living with rare NR2F1 mutations through education, awareness and research. As a result of joining forces with other foundations, we aim to be leaders in patient empowerment and patient-led research for the rare disease community at large ultimately serving as a model to other organizations.
Malan Syndrome Foundation is represented by Dr. Christal Delagrammatikas. The mission of the Malan Syndrome Foundation is to improve the lives of individuals and families affected by Malan syndrome in the global community through support, outreach and research. The Malan Syndrome Foundation is a volunteer, parent-led organization.
KIF1A.ORG is represented by Kathryn Atchely, President. KIF1A.ORG is a global community dedicated to improving the lives of those affected by KIF1A Associated Neurological Disorder and accelerating research to find a cure. Our relentless community of families, researchers, clinicians, innovators and supporters are determined to bring treatment to this generation of people affected by KAND.
GRIN2B Foundation is represented by Liz Marfia Ash, President & Founder. GRIN2B Foundation is a parent-run organization dedicated to furthering research on the GRIN2B gene and providing support and education to the small, but growing community of individuals and families impacted by a GRIN2B diagnosis. Though GRIN2B Foundation was the first GRIN2B-related organization formed, we are very proud to work in collaboration with many other GRIN2B and GRIN Disorder organizations that have since formed worldwide.
Glut1 Deficiency Foundation is represented by Glenna Steele, Executive Director. The Glut1 Deficiency Foundation is a nonprofit patient advocacy organization dedicated to improving lives in the Glut1 Deficiency community through its mission of increased awareness, improved education, advocacy for patients and families, and support and funding for research. We are working hard to bring help and hope to the Glut1 Deficiency community.
FOXG1 Research Foundation is represented by Nasha Fitter, CEO, Head of Research. The mission of the FOXG1 Research Foundation to accelerate research to find a cure for FOXG1 syndrome. We are dedicated to funding the world’s leading scientists that are integral along the Path to a Cure for all children with FOXG1 syndrome. We will continue to apply our research to solve related brain disorders.
Foundation for USP7 Related Diseases is represented by Bo Bigelow, Chairman/Co-Founder. Their mission is to cure Hao-Fountain Syndrome (previously known as USP7-related diseases). We do this by funding research and identifying more patients. In funding research, we seek to (1) uncover methods of activating USP7 to rescue this haploinsufficient phenotype; and (2) understand how alterations in proper functioning of endosomal protein recycling cause seizures and other neurological problems.
FamilieSCN2A Foundation is represented by Leah Schust Myers, Executive Director. Their vision is to find effective treatments and a cure for SCN2A related disorders. Their mission is to improve the lives of those affected by SCN2A related disorders through research, public awareness, family support and patient advocacy. FamilieSCN2A Foundation was created by parents of children suffering from SCN2A related disorders who work unwaveringly to support both families and research.
CureGPX4 is represented by Sanath Kumar Ramesh, Founder. The organization’s mission is to create treatments to Spondylometaphyseal Dysplasia Sedaghatian type (SSMD).
CureSHANK is represented by Geraldine Bliss, Founder and President. Their mission is to accelerate the development of treatments for Phelan-McDermid Syndrome and SHANK-related disorders. Their approach is to identify and fund projects that overcome critical barriers to successful drug development and to coordinate scientific efforts to improve efficiency and speed in the field.
CureGRIN is represented by Keith McArthur, CEO and Head of Science. Their mission is to improve the lives of people living with GRIN Disorder. The foundation is founded and run by parents that are committed to improve the lives of people with GRIN disorder.
CHAMP1 Research Foundation is represented by Jeff D’Angelo, Founder, Research Committee Chair. Their mission is to improve the lives of those affected by CHAMP1 through clinical research, effective treatments, public awareness, early detection, family support and patient advocacy.
CACNA1A Foundation is represented by Lisa Manaster, President. Their mission is to increase awareness of CACNA1A variants, support individuals and families affected by CACNA1A, and raise funds to support research and treatment options to find a cure for CACNA1A.
Share this article and raise awareness of Schinzel-Giedion Syndrome.
Newsletter Signup
Sign-up to receive family stories and updates on our research projects and fundraising campaigns.